
Stability of Fe(111)/Al2O3(0001) interface based on first principles
-
摘要:
稀贵金属催化剂催化甲烷分解效果显著但成本较高,为此,探寻低成本催化剂是实现其工业化推广的重要课题。铁基催化剂具有耐高温、价格低、性能好等优点,目前针对铁基催化剂的表面机理研究较少,催化剂界面组成及结构稳定性仍没有统一的观点。本研究基于密度泛函理论(DFT),利用第一性原理计算表面能、态密度、差分电荷密度,研究Fe(111)/Al2O3(0001)界面稳定性,并探讨不同端面的催化剂形成机制。结果表明:O端界面的吸附能和电荷浓度大于Al1和Al2端界面,O原子周围电荷聚集明显;Al1和Al2端界面的Fe和Al之间形成微弱金属键,而O端界面的Fe和Al 之间形成共价键、金属键和离子键,O端共价键强度高于Al1和Al2端界面。因此,O端Fe/Al2O3催化剂相比于其他端面结合更加紧密,同时表现出更理想的催化效果,上述研究结果为制备Fe/Al2O3催化剂及研究其界面的形成机制提供理论基础。
Abstract:Rare and precious metal catalysts have remarkable effects on methane decomposition, but the cost is high, so that it is an important task to explore low-cost catalysts to realize their industrialization and popularization. Iron-based catalysts have the advantages of high-temperature resistance, low price and good performance. At present, there is little research on the surface mechanism of iron-based catalysts, and there is still no unified view of catalyst interface composition and structural stability. In this study, based on density functional theory (DFT), the stability of the Fe (111) /Al2O3 (0001) interface was studied by first principles, and the formation mechanism of the catalyst with different end faces was discussed. The results of surface energy, state density and differential charge density analysis showed that the adsorption energy and charge concentration at the O-terminal interface were greater than those at the interface of Al1 and Al2, and the charge accumulation around the O atom was obvious. Weak metal bonds were formed between Fe and Al at the interface of Al1 and Al2, while covalent, metallic and ionic bonds were formed between Fe and Al at the interface of the O terminal. The O terminal covalent bond strength was also higher than the Al1 and Al2 terminal interfaces. Therefore, the O-end Fe/Al2O3 catalyst was more tightly bound than other end faces, and would also show a better catalytic effect. The above research results provide a theoretical basis for preparing the interface formation mechanism of Fe/Al2O3 catalyst.
-
0 引言
加压浸出是一种清洁的湿法冶金新技术, 具有浸取高效﹑流程短﹑无污染等优点[1], 在复杂硫化铜矿浸出方面具有广阔的应用前景[2-4].但复杂硫化铜矿在硫酸浸出过程中易“钝化”[5], 受该问题局限, 复杂硫化铜矿加压浸出目前仅停留在实验室研究阶段.
为克服矿石浸出“钝化”问题, 可以采用微波预处理方法[6-9].当微波快速加热矿物时, 由于微波穿过金属矿物产生很大的热应力, 致使矿物边缘出现微小裂缝[10], 这有可能是使矿物活性增强的原因之一.另外, 在微波场中, 大多数硫化矿(如黄铁矿、黄铜矿等)都能吸收微波能[11].物质吸收微波后, 其晶体结构、表面性质等有可能发生显著改变[12], 如黄铁矿在一定条件下经微波处理后会转变成磁黄铁矿[13].实践证明, 经微波活化, 原本难以浸出的铜精矿在中等温度(453 K)条件下即可顺利浸出[14].
为进一步完善微波活化铜精矿加压浸出工艺, 加强浸出行为及浸出过程研究是非常必要的.本文即以微波活化铜精矿为研究对象, 探讨微波活化铜精矿在408~453 K温度范围内加压浸出动力学, 进而分析铜、锌浸出过程的动力学控制因素.
1 实验
1.1 原料与仪器
实验所用铜精矿XRD谱图见图 1.由图 1可知, 铜精矿中主要的金属矿物有闪锌矿、黄铜矿、黝铜矿、黄铁矿、方铅矿.铜精矿粒度为-44 μm占99.7 %.铜精矿在微波功率82 W、每批处理量95 g条件下辐照时间120 s, 过程中未见铅、锌、硫、砷的挥发损失, 微波活化前后铜精矿主要元素成分见表 1.
表 1 微波活化前后铜精矿的主要元素成分 /%
硫酸及其他化学试剂均为分析纯.微波活化铜精矿加压浸出过程中通入工业纯氧.
主要实验仪器:G70D20ASP-DF型微波炉(广东格兰仕微波炉电器有限公司), 额定微波频率2450 MHz, 额定输出功率700 W; CJF-T型高压釜(大连通达反应釜厂), 内容积1.0 L.
1.2 实验过程
铜精矿经微波活化预处理后按一定液固比与浸出剂混合加入高压釜的钛胆中.高压釜加盖密封, 通过控制箱设定加热温度.升温至设定温度时, 向高压釜内通入工业纯氧并开始计时.浸出结束后通水冷却, 待釜内压力降为0后开启高压釜, 分别将浸出液和浸出渣送样分析.铜、锌浸出率计算均按渣计.
为保证各实验点间具有可比性, 控制每次实验时高压釜升温速率及降温速率相同.
为使浸出过程中, 反应物(即硫酸)浓度保持相对恒定, 在微波活化铜精矿加压浸出动力学实验中, 选取了相对偏高的初始硫酸浓度(1.23 mol/L)以及较大的液固比(mL/g)30/1.基于大量实验可知, 当搅拌转速达到500 r/min以上时, 可以保证1.0 L釜内气、液、固三相充分混合和氧的传质, 故在各次实验中控制搅拌转速恒定为500 r/min.本研究未考虑氧分压对微波活化铜精矿加压浸出动力学的影响, 各次实验氧分压恒定为0.6 MPa.
2 结果与讨论
2.1 铜的浸出速率
在408~453 K温度范围内, 铜浸出率随浸出时间变化关系如图 2所示.图 2中直线为铜浸出率在一定浸出时间范围内的拟合直线.由图 2可见, 在铜浸出达到平衡之前, 铜浸出率与浸出时间之间呈良好的直线关系, 相关系数R≥0.99.进一步由图 2可知, 对于各浸出温度而言, 在浸出达到平衡之前, 铜浸出率随浸出时间延长而不断增大的规律是基本一致的.当浸出温度升高至453 K时, 铜浸出达到平衡的时间缩短至150 min.与未经微波活化预处理的铜精矿加压浸出行为[15]比较可知, 微波活化前后, 铜浸出行为规律基本一致.
铜精矿微波活化前后, 铜浸出率对应浸出时间的拟合直线方程见表 2.由表 2可见, 在408~438 K温度范围内, 微波活化前后, 铜浸出速率未见明显变化, 当温度升高至453 K时, 铜浸出速率较活化前略有增大.为保证铜的高效浸出, 微波活化铜精矿浸出温度宜选择453 K.
表 2 铜精矿铜浸出率与浸出时间对应的直线关系
2.2 锌的浸出速率
在408~453 K温度范围内, 锌浸出率随浸出时间变化关系如图 3所示.图 3中直线为锌浸出率在一定浸出时间范围内的拟合直线.由图 3可见, 在锌浸出达到平衡之前, 锌浸出率与浸出时间之间呈良好的直线关系, 相关系数R≥0.98.进一步由图 3可见, 锌浸出率随浸出时间变化的规律同铜浸出.当温度升高至453 K时, 锌浸出达到平衡的时间缩至120 min.相对于铜浸出而言, 锌浸出更早达到平衡.与铜浸出类似, 铜精矿微波活化前后锌浸出行为规律也基本一致.
铜精矿微波活化前后, 锌浸出率对应浸出时间的拟合直线方程见表 3.由表 3可见, 微波活化前后, 锌浸出速率随浸出温度的变化规律是相同的, 即在408~438 K温度范围内, 随浸出温度升高, 锌浸出速率不断增大; 当浸出温度进一步升高至453 K后, 锌浸出速率不再明显变化.当浸出温度低于423 K时, 锌浸出速率较活化前略有增大; 而当浸出温度高于438 K后, 虽然锌浸出率高于微波活化前, 但锌浸出过程反而略有放缓.
表 3 铜精矿锌浸出率与浸出时间对应的直线关系
进一步比较表 2、表 3结果可知, 在408~438 K温度范围内, 锌浸出速率始终高于铜浸出速率, 锌浸出优先于铜; 而当温度进一步升高至453 K后, 铜浸出得以明显促进, 铜浸出优先于锌.上述规律同精矿微波活化前.
2.3 浸出过程控制性步骤
在铜精矿各金属硫化矿氧压酸浸过程中, 将有单质硫固体产物生成, 浸出总反应式如下:

(1) 因此, 反应式(1)可以用收缩核未反应模型来描述, 该模型同样适用于微波活化铜精矿.浸出控制步骤可分为液体边界层扩散控制、界面化学反应控制、固体产物层扩散控制[16].在加压浸出条件下, 因搅拌在气泡的作用下被强化, 所以浸出速率可不受液体边界层扩散控制.
经计算可知, 在408~453 K温度范围内, 对于微波活化铜精矿铜、锌浸出而言, 只有界面化学反应控制的动力学曲线才呈现良好的线性关系, 结果如图 4、图 5所示.1-(1-t)1/3~t之间拟合所得线性回归方程分别见表 4和表 5.
表 4 微波活化铜精矿铜浸出动力学方程 表 5 微波活化铜精矿锌浸出动力学方程
表 5 微波活化铜精矿锌浸出动力学方程
进一步由表 4、表 5所示的铜、锌浸出表观速率常数(k)求得对应不同浸出温度的lnk(如表 6、表 7所示).进一步以lnk对1/T作图, 如图 6、图 7所示, 无论对于铜还是锌浸出而言, lnk与1/T之间均呈良好的线性关系(相关系数R≥0.97).对于铜浸出而言, 所得直线斜率为-6798.88;对于锌浸出而言, 所得直线斜率为-5985.74.根据阿仑尼乌斯(Arrhenius)公式, 可求解浸出反应的活化能:
表 6 微波活化铜精矿铜浸出表观速率常数 表 7 微波活化铜精矿锌浸出表观速率常数
表 7 微波活化铜精矿锌浸出表观速率常数
对于铜浸出而言:

对于锌浸出而言:

计算所得表观活化能均处于40~300 kJ/mol范围内, 由此可见, 微波活化铜精矿加压浸出过程的确受界面化学反应控制, 浸出控制步骤同微波活化前.铜、锌浸出反应表观活化能较微波活化前有明显降低, 这说明, 铜精矿经微波活化后铜、锌浸出过程得以促进.
3 结论
(1) 在408~453 K范围内, 在铜、锌浸出达到平衡之前, 浸出率与浸出时间呈良好的线性关系.微波活化前后, 铜精矿铜、锌浸出行为规律基本一致.
(2) 在408~438 K温度范围内, 铜精矿微波活化前后, 铜浸出速率未见明显变化, 当温度升高至453 K后, 铜浸出速率较活化前略有增大.当温度低于423 K时, 锌浸出速率较活化前略有增大; 当温度高于438 K时, 锌浸出过程反而略有放缓.为保证铜的高效浸出, 微波活化铜精矿浸出温度宜选择453 K.
(3) 在408~438 K温度范围内, 锌浸出速率始终高于铜, 锌浸出优先于铜; 而当温度进一步升高至453 K后, 铜浸出优先于锌.上述规律同精矿微波活化前一致.
(4) 在408 ~453 K范围内, 微波活化铜精矿铜、锌浸出反应表观活化能分别为56.33 kJ/mol和49.77 kJ/mol, 铜、锌浸出过程受界面化学反应控制, 浸出控制步骤同微波活化前.经微波活化后铜精矿铜、锌浸出过程得以促进.
朱冬梅 -
![]()
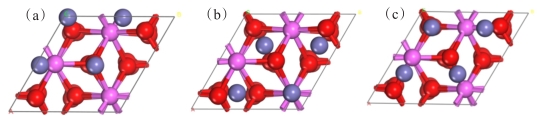
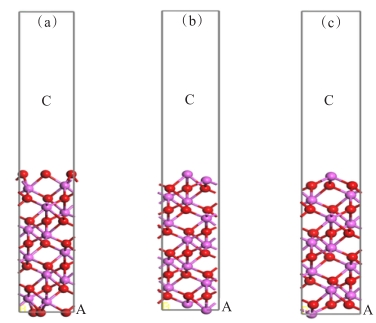
图 1 Al2O3(0001)结构优化后的表面模型,红色为氧原子,粉色为铝原子:(a)O端面;(b)Al2端面;(c)Al1端面
(a) O end face; (b) Al2 end face; (c) Al1 end face
Fig 1. Surface model of Al2O3(0001) after structure optimization, oxygen is red, and pink is aluminum:
![]()
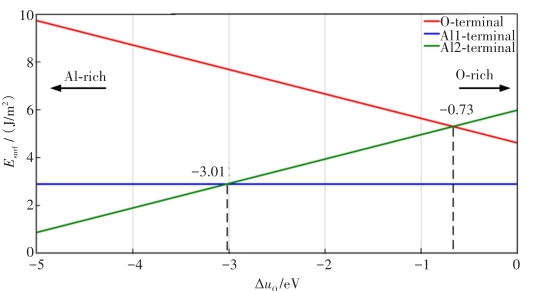
图 2 Al2O3(0001)的表面能与O化学势ΔuO之间的关系
Fig 2. Relationship between the surface energy of Al2O3 (0001) and the O chemical potential ΔuO
![]()
图 3 Fe(111)/Al2O3(0001)模型的俯视图:(a)Top位;(b)Hcp点位;(c)Bridge点位
Fig 3. Top view of Fe(111)/Al2O3(0001) models with different stacking sites:(a) Top-site; (b) Hcp-site; (c) Bridge-site
![]()
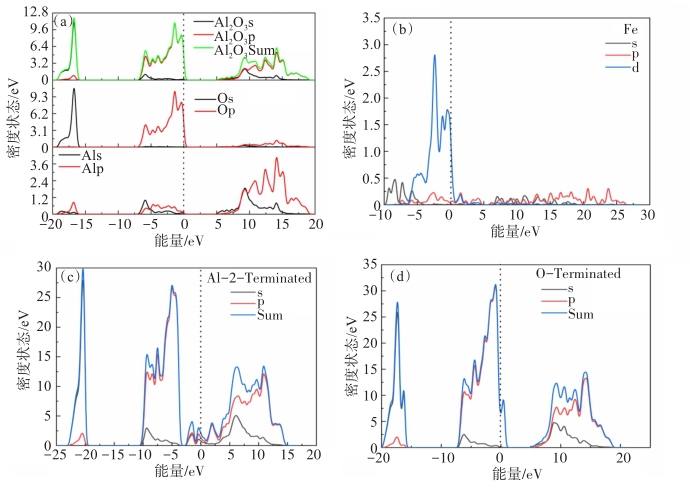
图 4 氧化铝的总态密度(DOS)和部分态密度(PDOS):(a)氧化铝;(b)铁;(c)Al2端氧化铝;(d)O端氧化铝
Fig 4. Total state density (DOS) and partial state density (PDOS) of (a) Alumina; (b) Fe; (c) Al2-terminal alumina; (d) O-terminal alumina
![]()
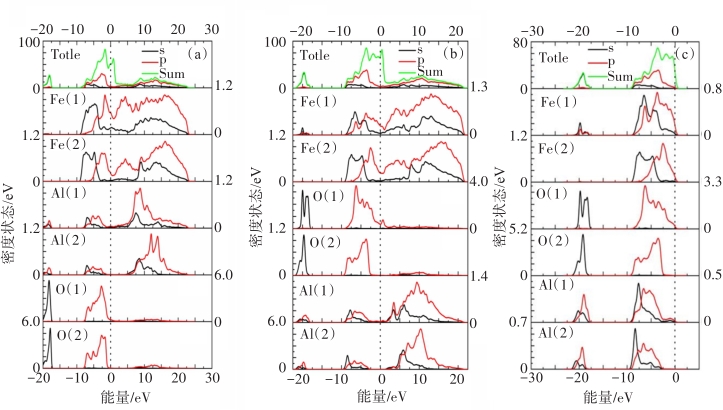
图 5 Fe(111)/Al2O3(0001)界面不同端面的PDOS:(a)Al1端界面;(b)Al2端界面;(c)O端界面
Fig 5. PDOS diagram of the different end faces of the Fe (111)/Al2O3(0001) interface:(a) the A11-end interface; (b) the Al2-end interface; (c) the O-end interface
![]()
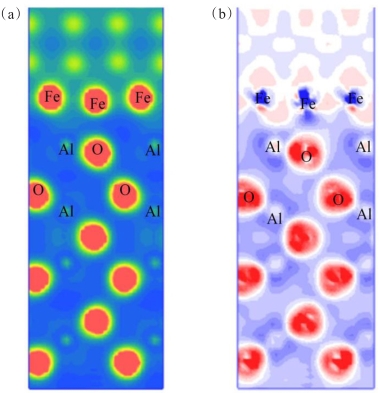
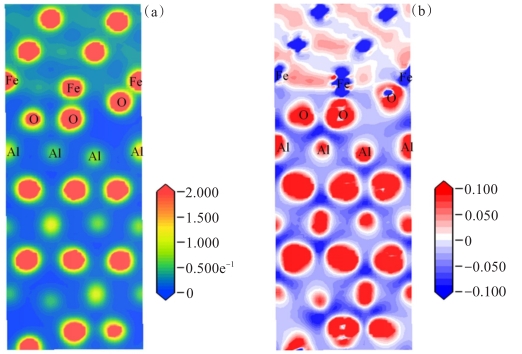
图 6 (a)Al端界面电荷密度;(b)Al端界面差分电荷密度
Fig 6. (a) Al terminal charge density; (b) Al terminal interface differential charge density
![]()
图 7 (a)O端界面电荷密度;(b)O端界面差分电荷密度
Fig 7. (a) O terminal charge density; (b) O terminal interface differential charge density
表 2 弛豫后不同Fe(111)/Al2O3(0001)结构的界面间距(d0)和吸附能(Wad)
Table 2 Interface spacing (d0) and adsorption energy (Wad) of different Fe(111)/Al2O3(0001) structures after relaxation
端面 堆积位置 d0(弛豫前)/Å 弛豫后 d0/Å Wad/(J/m2) O Top 1.8 1.69 7.21 Hcp 1.68 7.08 Bridge 1.71 6.93 Al1 Top 2.2 2.29 2.27 Hcp 2.21 2.47 Bridge 2.25 2.28 Al2 Top 2.0 2.13 2.87 Hcp 2.16 2.51 Bridge 2.22 2.41  下载: 导出CSV
下载: 导出CSV
-
[1] JIANG Z, LI L, XU J, et al. Density functional periodic study of the dehydrogenation of methane on Pd(111) surface[J]. Applied Surface Science, 2013, 286: 115-120.
[2] 卢立新, 王新东. 石墨烯负载铂催化剂的制备及稳定性[J]. 有色金属科学与工程,2015, 6(3):40-44. [3] ROBINSON A M, GIN M E, YUNG M M. Methane steam reforming kinetics on a Ni/Mg/K/Al2O3 Catalyst[J]. Topics In Catalysis, 2013, 56: 1708-1715.
[4] 张扬, 王韬翔, 刘贝, 等. 基于非晶态 Fe/Al2O3 催化剂的乙烷催化裂解高效制备碳纳米管[J]. 石油学报 (石油加工), 2023, 39(4): 750-759. [5] BAYAT N, REZAEI M, MESHKANI F. Methane decomposition over Ni-Fe/Al2O3 catalysts for production of COx-free hydrogen and carbon nanofiber[J]. International Journal of Hydrogen Energy, 2016, 41(3): 1574-1584.
[6] JANG H T, CHA W S. Hydrogen production by the thermocatalytic decomposition of methane in a fluidized bed reactor[J]. KOREAN J CHEM ENG, 2007, 24: 374-377.
[7] ZHOU L, ENAKONDA L R, SAIH Y, et al. Catalytic methane decomposition over Fe-Al2O3[J]. CHEM SUS CHEM, 2016, 9(11): 1243-1248.
[8] IBRAHIM A A, FAKEEHA A H, AL-FATESH A S, et al. Methane decomposition over iron catalyst for hydrogen production[J]. International Journal of Hydrogen Energy, 2015, 40(24): 7593-7600.
[9] LIU S, ZHANG X. Chemical concepts from density functional theory[J]. ACTA PHYS CHIM SIN, 2018, 34: 563-566.
[10] BECKE A D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction[J]. The Journal of Chemical Physics, 1992, 96(3): 2155-2160.
[11] FISCHER T H, ALMLOF J. General methods for geometry and wave function optimization[J]. The Journal of Physical Chemistry, 1992, 96(24): 9768-9774.
[12] YU J, LIN X, WANG J, et al. First-principles study of the relaxation and energy of bcc-Fe, fcc-Fe and AISI-304 stainless steel surfaces[J]. Applied Surface Science, 2009, 255(22): 9032-9039.
[13] 李立清, 周润, 龙慧婷, 等. 基于密度泛函理论的氢氧化镁(101)表面改性机理研究[J]. 有色金属科学与工程, 2023, 14(4): 447-453. [14] DONG N, ZHANG C, LIU H, et al. Effects of different alloying additives X (X= Si, Al, V, Ti, Mo, W, Nb, Y) on the adhesive behavior of Fe/Cr2O3 interfaces: A first-principles study[J]. Computational Materials Science, 2015, 109: 293-299.
[15] HAN C, ZHANG C, LIU X, et al. Effects of alloying on oxidation and dissolution corrosion of the surface of γ-Fe (111): a DFT study[J]. Journal of Molecular Modeling, 2015, 21: 1-9.
[16] CHEN L, LI Y, XIAO B, et al. First-principles calculation on the adhesion strength, fracture mechanism, interfacial bonding of the NiTi (111)//α-Al2O3 (0001) interfaces[J]. Materials & Design, 2019, 183: 108119.
[17] RUPPI S. Deposition, microstructure and properties of texture-controlled CVD α-Al2O3 coatings[J]. International Journal of Refractory Metals and Hard Materials, 2005, 23(4/5/6): 306-316.
[18] CHEN Y J, HOU C J, YANG Y. Surface energy and surface stability of cesium tin halide perovskites: a theoretical investigation[J]. Physical Chemistry Chemical Physics, 2023, 25(15): 10583-10590.
[19] DANG D Y, SHI L Y, FAN J L, et al. First-principles study of W-TiC interface cohesion[J]. Surface and Coatings Technology, 2015, 276: 602-605.
[20] LI R, CHEN Q, ZHANG Y, et al. Insight into diffusion-rebonding of Nano-Al2O3 on Fe surface in high-temperature thermal energy storage system[J]. Applied Surface Science, 2020, 530: 147249.
[21] LI R, CHEN Q, OUYANG L, et al. Adhesion strength and bonding mechanism of γ-Fe (111)/α-Al2O3 (0001) interfaces with different terminations[J]. Journal of Alloys and Compounds, 2021, 870: 159529.
[22] LI R, CHEN Q, ZHANG Z, et al. Revealing the atomic-scale structure and the fracture mechanism of the α-Al2O3/γ-Fe ceramic-metal interface[J]. Journal of Alloys and Compounds, 2021, 885: 161163.
[23] WANG Y, LIU X, YANG Q, et al. First principles calculation ofinterfacial stability, energy, and elemental diffusional stabilityof Fe (111)/A12O3 (0001) interface[J]. AlP Advances, 2019, 9(12): 125313.
[24] LI R, CHEN Q, OUYANG L, et al. Insight into the strengthening mechanism of α-Al2O3/γ-Fe ceramic-metal interface doped with Cr, Ni, Mg, and Ti[J]. Ceramics International, 2021, 47(16): 22810-22820.
[25] YANG H X, CHSHIEV M, DIENY B, et al. First-principles investigation of the very large perpendicular magnetic anisotropy at Fe| MgO and Co| MgO interfaces[J]. Physical Review B, 2011, 84(5): 054401.







计量
- 文章访问数: 116
- HTML全文浏览量: 29
- PDF下载量: 32
