
Morphology modulation of tungsten carbide and its electrochemical performance as a Pt cocatalyst
-
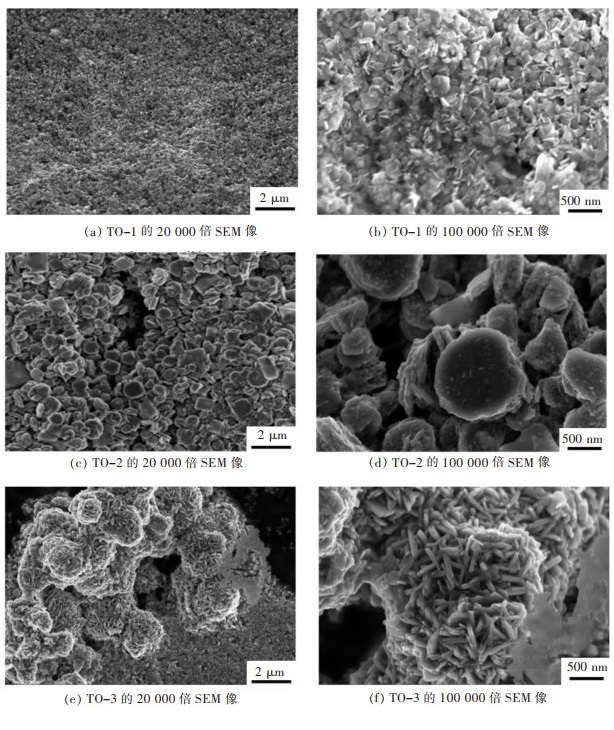
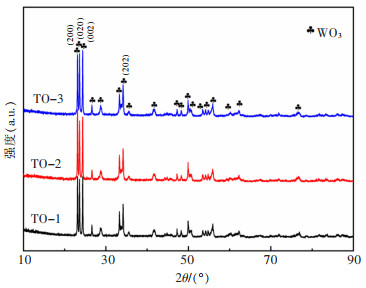
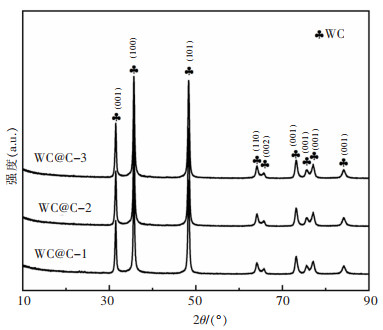
摘要: 本文采用水热模板法,通过添加表面活化剂柠檬酸和模板剂氧化石墨烯(GO)调控前驱体的形貌尺寸。将前驱体置于空气气氛中进行热处理,得到厚度薄、尺寸小、分散的WO3。在氮气保护气氛下,通入乙醇/甲醇液体碳源,并处于1 000 ℃下保温2 h,进行原位还原碳化反应,得到表面包覆石墨化碳层的碳化钨(WC@C)。在WC@C表面原位负载不同浓度的Pt NPs,得到Pt-WC@C复合催化剂。该催化剂Pt-WC@C-1(10% Pt)在碱性溶液中性能较佳,电流密度为10 mA/cm2下的过电势(η10)为187 mV,塔菲尔斜率为115.7 mV/dec(10% Pt);在酸性溶液中具有更优良的析氢催化性能,Pt-WC@C-2(5% Pt)性能优于Pt-WC@C-1(10% Pt)和Pt-WC@C-3(1% Pt),η10为87.6 mV,塔菲尔斜率为51.2 mV/dec。这主要归因于分散性较好的WC表面包覆了较薄的石墨化碳层。Abstract: In this paper, the hydrothermal template method was applied to regulate the morphology and size of the precursors by adding the surfactant citric acid and the template graphene oxide (GO). The precursor was placed in the air atmosphere for heat treatment to obtain thin, small-sized and dispersed WO3. Then, under a nitrogen protection atmosphere, the ethanol/methanol liquid carbon source was introduced and kept warm at 1 000 ℃ for 2 h to carry out the in-situ reduction carbonization reactions and the tungsten carbide (WC@C) coated with graphified carbon layer was obtained. Different concentrations of Pt NPs in situ at WC@C surface loading yielded a Pt-WC@C compound catalyst. The catalyst Pt-WC@C-1 (10% Pt) has the best performance in an alkaline solution, with an overpotential (η10) of 187 mV and a Taffir slope of 115.7 mV / dec (10% Pt) at the current density of 10 mA/cm2. In acid solution, the Pt-WC@C compound catalyst has a better performance in catalytic properties of hydrogen precipitation. The performance of Pt-WC@C-2 (5% Pt) is better than that of Pt-WC@C-1 (10% Pt) and Pt-WC@C-3 (1% Pt) with an overpotential (η10) of 87.6 mV and the Taffir slope of 51.2 mV/dec, which is mainly due to thinner graphitized carbon layers coated on the surface of well-dispersed WC particles.
-
Keywords:
- flake /
- tungsten carbide /
- graphene /
- catalyst /
- hydrogen evolution
-
分子动力学模拟即为Molecular Dynamics Simulation,简称MD[1~6],是计算复杂分子体系的有效方法.20世纪60年代Alder[7]等首先采用分子动力学模拟方法研究了凝聚态液体和气体的状态方程,开创了分子动力学研究物质宏观性质的先例.由于具有节省试验成本和时间,增加试验的安全性和提高试验的针对性等特点,所以经过多年的发展,分子动力学模拟方法目前已经拓展到了生命科学[8]、生物工程[9]、地球物理[10]、材料[11]、医学[12]等各个领域,已成为一种非常普遍的研究手段.
1 分子动力学模拟计算的组成及其原理
1.1 力场
分子动力学模拟是基于应用力场和牛顿运动力学方程所发展起来的一种计算机模拟方法[13].力场,是分子动力学模拟能够有效实现的基础.没有针对模拟体系合适的力场,就不可能得到正确的分子动力学模拟结果.
分子的总能量为动能与势能之和.分子的势能通常可表示为简单的几何坐标函数,以这样的数学形式表示的势能函数就称作力场.比如,将双原子分子AB的振动势能用A与B间键长的函数表示:
$$V(r)=\frac{1}{2}k{{(r-{{r}^{0}})}^{2}}$$ 其中,为弹力常数,为键长,为AB的平衡键长.此时,函数就称作双分子体系AB的力场.对于复杂体系的分子而言,其总势能一般表示为:
$$U={{U}_{nb}}+{{U}_{b}}+{{U}_{o}}+{{U}_{\phi }}+{{U}_{\chi }}+{{U}_{cl}}$$ 其中, ${{U}_{nb}}$ 为范德华非键结势能, ${{U}_{b}}$ 为键伸缩势能, ${{U}_{o}}$ 为键角弯曲势能, ${{U}_{\phi }}$ 为双面角扭曲势能, ${{U}_{\chi }}$ 为离平面振动势能, ${{U}_{cl}}$ 为库伦静电势能.
近几十年来,随着计算机硬件水平的不断提高,分子力学与分子动力学理论的不断发展,分子动力学模拟系统由原先的单原子分子系统逐渐拓展到含有复杂多原子分子体系.各种新型力场层出不穷,有些力场已经越来越完善.但是每个力场都有自己的适用范围和局限性.本文主要介绍了Material Studio软件中常见的几个力场,如COMPASS[14-16]、pcff[17]、Dreiding[18].
COMPASS (Condensed-phase Optimized Molecular Potentials for Atomistic Simulation Studies)力场的出现标志着力场研究的突破.它是第一个使用从头算法的力场,能够精确的模拟和预测气相的结构、构象、振动等性质,凝聚态的状态方程、内聚能等性质.正是由于COMPASS力场的大多数参数来源于从头算法,使它能够广泛地适用于共价分子体系,如大多数常见有机物、无机物和聚合物、金属、金属氧化物和金属卤化物.
pcff(polymer consistent forcefield)力场是基于CFF91力场发展而来的,被应用于聚合物和有机材料.pcff力场可用于聚碳酸酯类、多糖类等聚合物以及无机和有机材料,包括约20种金属(Li,K,Cr,Mo,W,Fe,Na,Ni,Pd,Pt,Cu,Ag,Au,Al,Sn,Pb)、糖类、脂类和核酸以及惰性气体(He,Ne,Kr,Xe).
Dreiding是一个万能力场,对预测有限原子的结构尤其擅长.Dreiding能够准确的预测大型分子结构,包括一些元素的奇异组合以及没有实验数据的分子结构.适用于计算有机、生物和主族无极分子的几何结构、构像能、分子间结合能和晶体堆积.
1.2 分子动力学的基本原理
分子动力学模拟与量子化学计算不一样,量子化学是以薛定谔方程为基础的,而分子动力学是以牛顿运动方程为基本原理.在含有N个分子的系统中,系统的总能量为内部分子的总动能和总势能之和.势能U是位置的函数,可分为分子间的相互作用力 ${{U}_{vdw}}$ (范德华作用力)以及分子内部的势能 ${{U}_{\operatorname{int}}}$ :
$$U={{U}_{vdw}}+{{U}_{\operatorname{int}}}$$ (1) 系统中总势能为系统内各原子坐标如键长的伸缩、键角的弯曲等势能的总和.
依据牛顿运动定律,计算系统中各原子所受力的加速度,可得到经过 $\delta t$ 后各分子的位置和速度.再由新的位置出发,重复以上步骤,计算各原子所受的力以及加速度,得到新的位置和速度.如此循环往复,就可得到一定时间后系统中各原子的位置和速度.然后通过位置和速度,计算系统的势能和动能,通过式(1) 就可以得到系统的总能量以及温度等宏观性质.
1.3 牛顿运动方程的数值解法
分子动力学计算中为了得到体系原子的速度和位置,必须求解牛顿运动方程.由于要考虑到截断误差、累积误差等不可避免的误差因素,因此合适的算法在解牛顿运动方程时就显得尤为重要,不正确的算法很可能得到错误的模拟结果.求解牛顿运动方程,需要将方程化为有限差分方程,通过有限差分方法来求解.经过几十年的发展,已经发展了很多种方法,这里主要介绍一种使用最普遍和可靠的算法:Verlet算法.
Velert方法是解牛顿运动方程最常用的方法.起初的Velert方法,是将原子的起始位置按照泰勒公式展开:
$$r\left( t+\delta t \right)=r\left( t \right)+\frac{d}{dt}r\left( t \right)\delta t+\frac{1}{2!}\frac{{{d}^{2}}}{d{{t}^{2}}}r\left( t \right){{\left( \delta t \right)}^{2}}+\cdots $$ (2) 将式(2) 中的变换符号为,得:
$$r\left( t-\delta t \right)=r\left( t \right)-\frac{d}{dt}r\left( t \right)\delta t+\frac{1}{2!}\frac{{{d}^{2}}}{d{{t}^{2}}}r\left( t \right){{\left( \delta t \right)}^{2}}+\cdots $$ (3) 将式(2) 与公式(3) 相加,得位移公式:
$$r\left( t+\delta t \right)=-r\left( t-\delta t \right)+2r\left( t \right)+\frac{{{d}^{2}}}{d{{t}^{2}}}r\left( t \right)\left( \delta t \right)$$ (4) 将式(3) 与(2) 相减,得速度公式:
$$v\left( t \right)=\frac{dr}{dt}=\frac{1}{2\delta t}\left[ r\left( t+\delta t \right)-r\left( t-\delta t \right) \right]$$ (5) 式(5) 的缺点在于假若 $\delta t$ 很小,则容易由 $\frac{1}{\delta t}$ 放大后引起误差.为了避免这种误差的出现,Velert发展了另外的一种差分计算公式——跳蛙方法.
跳蛙方法中描述速度与位置的数学公式为:
$$\begin{align} & \overrightarrow{{{v}_{i}}}\left( t+\frac{1}{2}\delta t \right)=\overrightarrow{{{v}_{i}}}\left( t-\frac{1}{2}\delta t \right)+\overrightarrow{{{a}_{i}}}\left( t \right)\delta t \\ & \overrightarrow{{{r}_{i}}}\left( t+\delta t \right)=\overrightarrow{{{r}_{i}}}\left( t \right)+\overrightarrow{{{v}_{i}}}\left( t+\frac{1}{2}\delta t \right)\delta t \\ \end{align}$$ (6) 在进行计算时假设 $\overrightarrow{{{v}_{i}}}\left( t-\frac{1}{2}\delta t \right)$ 与 $\overrightarrow{{{r}_{i}}}\left( t \right)$ 已知,则由时的位置 $\overrightarrow{{{r}_{i}}}\left( t \right)$ 计算质点所受的力和加速度 $\overrightarrow{{{a}_{i}}}\left( t \right)$ .再根据式(6) 推测在时间变化为 $\frac{1}{2}\delta t$ 时间内的速度 $\overrightarrow{{{v}_{i}}}\left( t+\frac{1}{2}\delta t \right)$ ,依此类推.时间为时的速度由式(7) 算出:
$$\overrightarrow{{{v}_{i}}}=\frac{1}{2}\left[ \overrightarrow{{{v}_{i}}}\left( t+\frac{1}{2}\delta t \right)+\overrightarrow{{{v}_{i}}}\left( t-\frac{1}{2}\delta t \right) \right]$$ (7) 利用Velert跳蛙方法计算只需保存 $\overrightarrow{{{v}_{i}}}\left( t-\frac{1}{2}\delta t \right)$ 和 $\overrightarrow{{{r}_{i}}}\left( t \right)$ 2种参数,可以节省存储空间.另外,这种方法的简便性和准确性使得Velert跳蛙方法得到广泛的应用.
1.4 系综
通过求解牛顿运动方程能够对一个系统进行分子动力学模拟,但是这个系统必须是一个能量恒定的、与外界无交流的系统.通常条件下,系统是暴露在外部环境中的,存在着与外部能量的相互交换.这样的系统,它的总能量不是一个定值.因此,对分子动力学模拟的适用条件进行扩展就显得尤为必要.系综[19-22]是一个统计学的概念,在不同的系综条件下,根据系统的原子数、压强、温度、体积、能量、焓等性质的变化就可以计算出体系的结构、能量和动力学性质.在这里介绍一个等温系综(NVT)和一个绝热系综(NVE).所有系综必须保持原子数量的恒定,其中,NPT和NPH系综只适用于周期性的系统中,因为非周期性的系统中体积是未定义的.
1.4.1 NVE系综
NVE系综是一个能量恒定、体积恒定的系综,也被称为微正则系综.它来源于求解牛顿运动方程时没有任何的温度和压力控制,处于绝热条件下的能量守恒.由于在求解积分方程过程中存在舍去和截断误差,所以能量值会有波动并偏离正常值.NVE系综适用于恒定能量的构象空间或者不产生温度和压力干扰的系统.NVE系综常用来计算热力学反应常数.
1.4.2 NVT系综
NVT系综是一个恒定温度和恒定体积的系综,也被称为正则系综,它是通过对热力学温度的控制得到的.为保持系统的温度不变,典型的控温方法是采用Nosé-Hoover热浴法.NVT系综适用于真空中无周期性边界条件下的模型构象搜索.由于没有耦合压力的影响,在周期性边界条件下,NVT系综具有对计算轨迹扰动很少的优势.
2 分子动力学模拟计算结果的分析
分子动力学模拟计算可以得到系统的运动轨迹、各种物理量的平均值以及热力学和动力学性质等许多有用信息[23-24].
2.1 轨迹分析
分子动力学模拟可以计算系统内每一个原子或者基团在每一段时间内的位置和速度,这些位置和速度随着时间的变化而变化,因此就形成了原子或者基团运动的途径,这些途径就称作轨迹.通常在进行动力学模拟计算前会对计算进行设置,以一定的时间间隔存取系统内的原子或基团的位置和速度信息,待到计算完以后,就可以调用这些信息进行热力、统计以及动态的轨迹分析.
2.2 温度
物理学上系统的温度可由系统内部原子的动能平均值而得到:
$$T=\frac{\left\langle 2K \right\rangle }{f}$$ 其中,K为动能,T为温度,f为系统的自由度.对含有N个原子的系统, $f=3N-3$ ;假如还有限制条件则每加一个限制条件减去一个自由度.
2.3 扩散系数
利用分子动力学模拟技术计算扩散系数的常用方法有2种,为Einstein关系式法和Green-Kubo方法.Einstein关系式法的公式为:
$$D=\frac{1}{6}\underset{t\to \infty }{\mathop{\lim }}\,\frac{d}{dt}\sum\limits_{i=1}^{N}{{{X}_{i}}}\left\langle {{\left| {{r}_{i}}(t)-{{r}_{i}}(0) \right|}^{2}} \right\rangle $$ Green-Kubo方法的计算公式为:
$$D=\frac{1}{3}\int_{0}^{\infty }{\left\langle {{v}_{i}}(t)-{{v}_{i}}(0) \right\rangle dt}$$ 对于经典体系,Einstein和Green-Kubo 2种方法严格等价.
此外,分子动力学模拟的结果除了可以对以上3个性质进行分析外,还可以对系统的密度、密度场、键长分布、径向分布、压力能量组成、X射线散射谱等性质进行分析.
3 分子动力学模拟在选矿中的应用
在选矿领域,为了探究浮选药剂对矿物作用的本质,通常采用接触角测量、浮选溶液pH值及温度变化、电性分析法、矿物表面酸碱性、溶液溶解作用法以及X射线光电子谱(XPS)、紫外光电子谱(UPS)、俄歇电子谱(AES)、二次离子质谱(SIMS)、红外光谱(IR)等各种光谱方法来开展研究.这些方法能够加强对矿物浮选机理的探究,但是却不能够将矿物与药剂作用的过程具体形象的展现给大家.随着科学技术的发展,特别是计算机科学的飞速提升,使得通过计算研究矿物与药剂的相互作用成为可能.特别是一些分子动力学模拟软件,比如Material Studio、AMBER、NAMD等软件的出现,使得矿物与药剂相互作用的过程可以通过计算机清晰的展现在人们面前.通过对矿物与药剂相互作用的分子动力学模拟,可以揭示药剂如何具体地作用于矿物表面;可以更好的了解药剂对矿物作用的影响;可以探究矿物本身的一些性质.
3.1 分子动力学模拟矿物浮选试验
通过对矿物表面与药剂相互作用的分子动力学模拟,能够提供在分子微观水平的药剂与矿物相互作用的情况,加深对药剂与矿物之间相互作用的理解,为实际矿石的浮选提供理论依据.
王振等[25]利用分子动力学模拟的方法研究了新型捕收剂氯化十六烷基吡啶(CPC)在氧化钼表面的吸附行为.首先建立了氧化钼、磷灰石的晶体模型,然后运用量子化学计算的方法对它们的晶体结构和表面性质进行了研究,同时优化建立好的药剂CPC分子,然后将药剂CPC与氧化钼(100) 面和磷灰石(010) 面分别在真空环境下进行分子动力学模拟,并对模拟结果进行分析,结果显示CPC与氧化钼(100) 面的相互作用能为-448.86 kJ/mol,要大于CPC与磷灰石(010) 面的相互作用能-420.16 kJ/mol,表明CPC更易与氧化钼表面相互作用,这与单矿物浮选实验以及红外光谱测试的结果相一致.
陈攀等[26]利用分子动力学模拟的方法研究了季盐在高岭石(001) 面上的吸附行为.实验构建了十二烷基三甲基氯化铵(DTAC)和十四烷基氯化(TTPC)的分子结构模型,经过100 ps的分子动力学模拟发现,DTAC分子几乎垂直作用于矿物表面,TTPC的头基的多条支链均稳稳的附着于矿物表面,TTPC与矿物表面接触面积要大于DTAC与矿物表面的接触面积,吸附能计算结果也显示TTPC与矿物表面的相互作用能(-809.222 kcal/mol)要大于DTAC与矿物表面的相互作用能(-353.323 kcal/mol),表明TTPC更易于吸附在高岭石表面,浮选表现更优异.
孙伟等[27]对钒云母的极完全解理面(001) 面与石英的(001) 面进行吸附捕收剂T的分子动力学模拟研究浮选机理.通过对分子动力学模拟后的结构进行吸附计算,得到捕收剂T与钒云母的吸附能为-47.83 kJ/mol,与石英表面的吸附能为-1.02 kJ/mol,证实了捕收剂T对钒云母的选择捕收作用.
3.2 分子动力学模拟揭示浮选过程中药剂的作用机理
通过对药剂浮选行为的分子动力学模拟,可以查明药剂分子在两相界面发生的相互作用行为,为药剂分子对矿物表面的附着作用提供理论指导.
王玉才[28]利用分子动力学模拟方法研究了油/水界面十六烷基邻二甲苯磺酸钠聚集的行为.通过构建十六烷基邻二甲苯磺酸钠的分子结构模型,运用量子化学软件GAMESS(US)对其进行了量子化学结构优化,将优化好的分子在油/水界面环境中进行动力学模拟.模拟结构表明,第一水层和第二水层的形成是由于磺酸基中的氧原子与水分子中的氢原子之间存在着氢键作用,而在第一水层与第二水层中存在着反离子Na+.磺酸基与水分子之间的氢键并不会因为表面活性剂分子数量的增加而改变.由于癸烷分子与十六烷基邻二甲苯磺酸钠之间表现出较强的亲和性,因此可以通过十六烷基邻二甲苯磺酸钠降低油水界面的张力.
徐前进[29]利用分子动力学模拟的方法研究了4种淀粉(SS、CMS、CAS和AMS)在一水硬铝石(010) 面和高岭石(001) 面的吸附行为.在真空环境下的分子动力学模拟研究结构表明,CMS和SS在一水硬铝石(010 )面上因为呈卧式吸附具有较大的投影面,而CAS和AMS在一水硬铝石(010) 面呈“L”式吸附具有较小的投影面,前者吸附层厚度分别为0.29 nm、0.57 nm,后者吸附层厚度分别为1.98 nm、2.02 nm.SS和CAS在高岭石(001) 面没有发生吸附行为,CMS和AMS在高岭石(001) 晶面只形成微弱的吸附作用.在水环境中对4种淀粉在一水硬铝石(010) 面的分子动力学模拟结果进一步证明了4种淀粉在一水硬铝石表面的吸附作用,WMS呈树状吸附于表面,WMS要比SS更易吸附在表面,而CMS由于大的取代度而占据多的吸附位点造成整体吸附量的下降.
3.3 分子动力学模拟对矿物自身结构的模拟研究
通过对矿物结构的分子动力学模拟研究,可以得出矿物结构之间存在的其它分子对其结构的影响,可为矿物结构因为外界条件的变化情况做出正确的判断.
况联飞等[30]应用分子动力学模拟对晶层间不同含水量的蒙脱土-水-离子体系和NaCl水溶液进行了研究,比较了水分子及离子特性和水溶液在矿物晶层间中的差别.分子动力学模拟计算结果表明蒙脱矿物同晶置换位置影响层间补偿阳离子,蒙脱矿中的四面体氧原子则与层间水分子形成配位氢键而与矿物表面定向排列;因为受到黏土矿物几何限制以及表面电荷的影响,水分子及离子在沿zz方向的扩散系数很小且要比NaCl水溶液小;径向分布函数的峰值强度表明晶层间水的高聚合度.分子动力学模拟结果为进一步研究黏土的高压力学特性奠定了基础.
全学波等[31]采用BMW-MARTINI粗粒化分子动力学模拟方法,对疏水性表面润湿状态受表面拓扑结构的影响进行了研究,研究结果表明润湿状态对润湿性表面的影响要比增大表面的粗糙度对润湿性的影响大.当柱间距小于4.7 nm时,柱间距和柱高对水珠在微柱结构疏水表面的润湿行为产生双重影响.在柱间距一定时,可以使水珠在表面的润湿状态超过一个临界的柱高由Wenzel态转变为Cassie-Baxter态,且润湿状态的转变与柱间距与柱高的比值有关,当比值小于2时,水珠层Wenzel态,当大于2时,水珠层为Cassie-Baxter态.能量分析结果表明水珠与表面的范德华作用决定着润湿状态的转变.
3.4 分子动力学模拟设计与筛选新型选矿药剂
随着选矿技术的不断进步,使得选矿工艺得到逐渐完善,不断进步,选矿设备也在不断改进,趋向于大型化高效化.但是,我国的选矿药剂研究与开发一直没有太大的起色,严重制约了选矿技术的发展.分子动力学模拟的出现为选矿药剂的研发提供了更好的平台.
杨刚等[32]通过分子动力学模拟软件计算了矿物表面的电子结构,并模拟了巯基苯并类浮选剂在浮选过程中的浮选行为,发现巯基苯并类浮选剂的p-π共轭结构在浮选过程中起着至关重要的作用,决定了捕收剂捕收效果的好坏.因此通过在BMT—的苯环上引入不同的取代基,如—CH3、—C6H5、—OCH3,来形成p-π共轭结构,指导新药剂的合成.研究发现,与苯环形成p-π共轭结构越强的基团所形成的新药剂,其捕收能力越强,选择性也 越好.并据此合成新型捕收剂5-苯基-2-巯基苯并噻唑(BNBT).经过对方铅矿的浮选实验发现,在矿浆浓度为1×10-5 mol/L时,浮选回收率可达到89 %.随着浓度的增加,回收率不断增大.浓度为1×10-4 mol/L时,回收率已经高达98 %,表现出了新药剂优越的捕收能力.
普拉蒂普等[33]通过分子动力学模拟评价新浮选药剂(烷基羟肟酸盐)的浮选效果.烷基羟肟酸分子经UFF优化,使用公式:
(△)=E络-[E药+E表]
E络、E药、E表分别为优化后的表面药剂络合物、药剂分子和表面群的总能量.
计算相互作用能.相互作用能的值越小,表明矿物表面和药剂的作用效果越好.通过计算得到烷基羟肟酸分子与氟碳铈矿的互相作用为-66.4 kcal/mol远小于氟碳铈矿与水的相互作用能-24.0 kcal/mol,表明该药剂对氟碳铈矿具有较好的作用效果.从大量的备选药剂中,筛选出最优的药剂分子结构,为高效新药剂的开发缩短了时间,节约了成本.
4 结束语
分子动力学具有传统选矿研究方法不具备的优势,为浮选行为研究,矿物与药剂作用机理揭示、矿石结构的探索和新型药剂分子的设计筛分等提供了一个更加简单、高效的模拟系统.分子动力学模拟必将是新型选矿技术研究的重要手段.然而,分子动力学模拟基于牛顿运动定律使用的是经验或半经验的方法,有其自身不完善的地方,在运用分子动力学模拟方法开展选矿研究时,要善于与传统的选矿研究方法相结合;对于其本身来讲,发展更好的力场,与量子化学更紧密的联系将是分子动力学发展的主要改进方向.相信将来分子动力学模拟方法会成为选矿研究的一项常规内容.
-
![]()
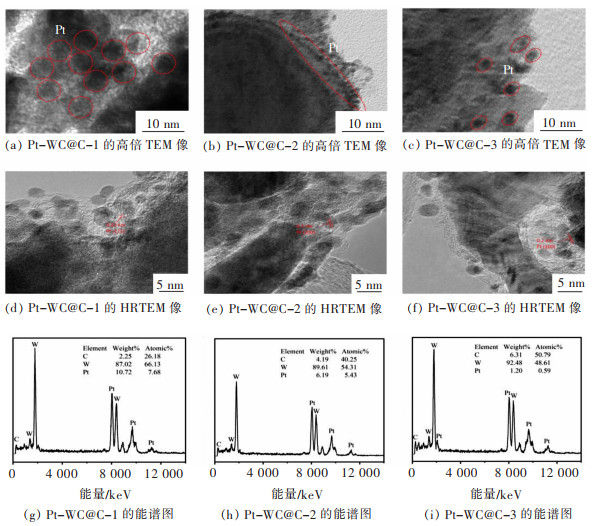
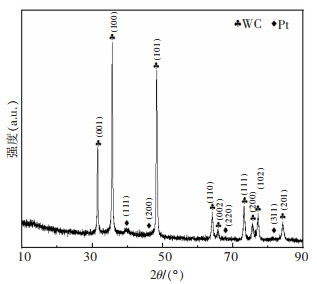
图 8 Pt-WC@C-1、Pt-WC@C-2和Pt-WC@C-3的TEM像和能谱图
Fig 8. TEM images of Pt-WC@C-1, Pt-WC@C-2 and Pt-WC@C-3
![]()
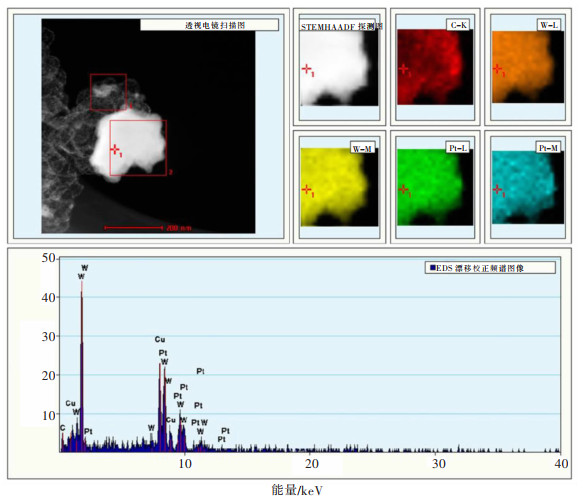
图 11 Pt-WC@C电极在1 mol/L KOH中扫描速率为10 mV/s的析氢极化曲线和塔菲尔斜率
Fig 11. HER polarization curves with a scan rate of 10 mV/s and corresponding Tafel plots of the Pt-WC@C electrode toward the HER in acidic electrolyte (1 mol/L KOH)
![]()
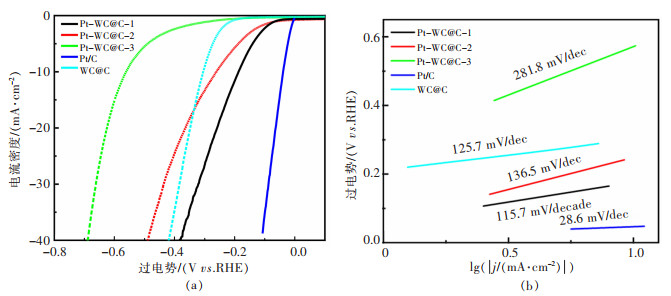
图 12 Pt-WC@C电极在0.5 mol/L H2SO4中扫描速率为10 mV/s的析氢极化曲线和塔菲尔斜率
Fig 12. HER polarization curves with a scan rate of 10 mV/s and corresponding Tafel plots of the Pt-WC@GCFs electrode toward the HER in acidic electrolyte (0.5 M H2SO4)
表 1 在酸性和碱性电解液条件下,Pt-WC@C复合材料对HER的电催化活性与其他形态的WC的比较
Table 1 Under acidic and alkaline electrolyte conditions, Pt-WC@C comparison of electrocatalytic activity of composite materials for HER with other WC forms

 下载: 导出CSV
下载: 导出CSV
-
[1] PENG X, NIE X, ZHANG L, et al. Carbon-coated tungsten oxide nanospheres triggering flexible electron transfer for efficient electrocatalytic oxidation of water and glucose[J]. ACS Applied Material Interfaces, 2020, 12 (51): 56943-56953. doi: 10.1021/acsami.0c13547
[2] ZHANG Z, LU N, YUAN Y, et al. Ultrafine cable-like WC/W2C heterojunction nanowires covered by graphitic carbon towards highly efficient electrocatalytic hydrogen evolution[J]. Journal of Materials Chemistry A, 2018, 6(31): 15395-15403. doi: 10.1039/C8TA05007D
[3] LING Y, YANG Z, ZHANG Q, et al. A self-template synthesis of defect-rich WS2 as a highly efficient electrocatalyst for the hydrogen evolution reaction[J]. Chemical Communications, 2018, 54: 2631-2634. doi: 10.1039/C7CC08962G
[4] ZHENG W Q, WANG L, DENG F, et al. Durable and self-hydrating tungsten carbide-based composite polymer electrolyte membrane fuel cells[J]. Nat. Commun., 2017, 8(1): 1-8. doi: 10.1038/s41467-016-0009-6
[5] CRUZ D C, CHRISTOFORO A L, SORDI V L, et al. Inducement of residual stresses in WC-5%Co cutting inserts by plunge-face grinding[J]. The International Journal of Advanced Manufacturing Technology, 2021, 113(1): 553-563.
[6] 朱奕松, 尹艳红, 刘开喜, 等. 钨源对碳化钨分散性及电催化性能的影响[J]. 有色金属科学与工程, 2017, 8(4): 73-79. doi: 10.13264/j.cnki.ysjskx.2017.04.013 [7] LU C, TRANCA D, ZHANG J, et al. Molybdenum carbide-embedded nitrogen-doped porous carbon nanosheets as electrocatalysts for water splitting in alkaline media[J]. ACS Nano 2017, 11(4): 3933-3942. doi: 10.1021/acsnano.7b00365
[8] HOU J, SUN Y, LI Z, et al. Electrical behavior and electron transfer modulation of nickel-copper nanoalloys confined in nickel-copper nitrides nanowires array encapsulated in nitrogen-doped carbon framework as robust bifunctional electrocatalyst for overall water splitting[J]. Advanced Functional Materials, 2018, 28(37): 1803278.1-183278.8.
[9] TOGHRAIE D, AGHAHDI M H, SINA N, et al. Application of artificial neural networks for predicting the viscosity of tungsten oxide (WO3)-MWCNTs/engine oil hybrid nanofluid[J]. International Journal of Thermophysics, 2020, 41(12): 1-17.
[10] 尹艳红, 吴子平, 赵曼, 等. 超细氧化钨的制备及其光催化性能研究[J]. 有色金属科学与工程, 2014, 5(3): 50-55. doi: 10.13264/j.cnki.ysjskx.2014.03.009 [11] LEVY R B, BOUDART M. Platinum-like behavior of tungsten carbide in surface catalysis[J]. Science, 1973, 181(4099): 547-549. doi: 10.1126/science.181.4099.547
[12] 尹艳红, 童珍, 幸康虔, 等. 以锯齿状碳纳米管为模板原位合成微/纳米球状氧化钨[J]. 稀有金属材料与工程, 2019, 48(1): 9-15. https://www.cnki.com.cn/Article/CJFDTOTAL-COSE201901002.htm [13] ESPOSTITO D V, HUNT S T, STOTTLEMYER A L, et al. Low-cost hydrogen-evolution catalysts based on monolayer platinum on tungsten monocarbide substrates[J]. Angewandte Chemie International Edition, 2010, 49(51): 9859-9862. doi: 10.1002/anie.201004718
[14] ESPOSTITO D V, CHEN J G, HUNT S T, et al. Monolayer platinum supported on tungsten carbides as low-cost electrocatalysts: opportunities and limitations[J]. Energy & Environmental Science, 2011, 4(10): 3900-3912.
[15] 张娜, 刘蕊, 聂晓荣, 等. 新型纳米碳/碳化钨的制备与应用研究[J]. 化工新型材料, 2020, 48(2): 250-253. https://www.cnki.com.cn/Article/CJFDTOTAL-HGXC202002054.htm [16] WANG X L, TANG Y J, HANG W, et al. Efficient electrocatalyst for the hydrogen evolution reaction derived from polyoxotungstate/ polypyrrole/graphene[J]. Chem Sus Chem, 2017, 10: 2402-2407. doi: 10.1002/cssc.201700276
[17] GONG Q, WANG Y, Hu Q, et al. Ultrasmall and phase-pure WC nanoparticles for efficient electrocatalytic and photoelectrochemical hydrogen evolution[J]. Nature Communications, 2016, 7: 13216.
[18] ZENG M, CHEN Y, LI J, et al. 2D WC single crystal embedded in graphene for enhancing hydrogen evolution reaction[J]. Nano Energy, 2017, 33: 356-362.
[19] REN B W, LI D Q, JIN Q Y, et al. Novel porous tungsten carbide hybrid nanowires on carbon cloth for high-performance hydrogen evolution[J]. Journal of Materials Chemistry A. 2017, 5(25): 13196-13203.
[20] LIU Z W, HOU X T, XI K, et al. Thickness controllable and mass produced WC@C@Pt hybrid for efficient hydrogen production[J]. Energy Storage Materials, 2018, 10: 268-274.
[21] HEO S, DAHLMAN C, STALLER C, et al. Enhanced coloration efficiency of electrochromic tungsten oxide nanorods by site selective occupation of sodiumions[J]. Nano Letters, 2020, 20(3): 2072-2079.
[22] EMIN S, ALTINKALA C, SEMERCI A, et al. Tungsten carbide electrocatalysts prepared from metallic tungsten nanoparticles for efficient hydrogen evolution[J]. Applied Catalysis B Environmental, 2018, 236: 147-153.
[23] HU Y, YU B, LI W X, et al. W2C nanodot-decorated CNT networks as a highly efficient and stable electrocatalyst for hydrogen evolution in acidic and alkaline media[J]. Nanoscale, 2019, 11(11): 4876-4884.
[24] CHEN Z, GONG W, CONG S, et al. Eutectoid-structured WC heterostructures: a new platform for long-term alkaline hydrogen evolution reaction at low overpotentials[J]. Nano Energy, 2019, 68: 104-335.
[25] TONG Z, WEN M, LV C, et al. Ultrathin and coiled carbon nanosheets as Pt carriers for high and stable electrocatalytic performance[J]. Applied Catalysis B: Environmental, 2020, 269: 118764-1-9.
[26] FAN X, ZHOU H, GUO X. WC nanocrystals grown on vertically aligned carbon nanotubes: an efficient and stable electrocatalyst for hydrogen evolution reaction[J]. ACS Nano, 2015, 9: 5125-5134.



计量
- 文章访问数: 182
- HTML全文浏览量: 36
- PDF下载量: 13
